Nova Diretriz de Hemorragia Digestiva Baixa
O Colégio Americano de Gastroenterologia (ACG) publicou em 2023 uma nova diretriz sobre hemorragia digestiva baixa [1]. Esse tópico resume as principais recomendações desse documento, que atualiza a última diretriz de 2016.
O que é e o que causa hemorragia digestiva baixa?
Hemorragia digestiva baixa (HDB) aguda é a saída de sangue pelo reto de origem colorretal. A incidência de HDB tem aumentado, provavelmente devido ao envelhecimento da população e ao maior uso de anticoagulantes e antiagregantes plaquetários.
A causa mais comum de HDB é a doença diverticular do cólon. Outras causas comuns são: colite isquêmica, hemorroidas, angiectasia, neoplasia colorretal, úlceras (estercorais, retais ou induzidas por AINEs) e colites (inflamatória, infecciosa ou relacionada a radioterapia).
O sangramento digestivo de intestino delgado é outra entidade e tem manejo diferente.

Registre-se e ganhe 2 tópicos gratuitos da sua preferência!
Se você já é um membro do Guia TdC, faça o Login. Faça o login e conheça o Guia!Veja o que o Guia TdC te oferece:
- O Guia TdC é uma biblioteca de estudos bem selecionados resumidos em texto e em áudio, juntamente com todas as referências utilizadas. Tudo isso por um preço super acessível.
- Receba semanalmente as últimas recomendações e novidades com impacto direto na prática diária.
- Junte-se aos mais de 3.000 assinantes e atualize-se sem esforço.
Como é o manejo inicial?
O exame físico inicial deve ser focado na avaliação de sinais de choque. A ressuscitação hemodinâmica com cristalóides deve ser realizada simultaneamente. O uso de ácido tranexâmico está contraindicado. Veja mais sobre o uso do ácido tranexâmico em "Ácido Tranexâmico no Peri-Operatório".
O exame do reto pode encontrar a fonte de sangramento nos casos das lesões anorretais ou indicar a presença de melena, sinal de hemorragia digestiva alta (HDA).
Quando o sangramento por uma HDA é intenso pode ocorrer hematoquezia. História de úlcera péptica, cirrose hepática e relação ureia/creatinina elevada (maior que 60) apontam para HDA. Se houver suspeita clínica de HDA, uma endoscopia digestiva alta deve ser realizada. A aspiração nasogástrica não tem boa sensibilidade e não está indicada para excluir HDA. Veja mais sobre HDA no episódio 120: caso clínico de hemorragia digestiva alta.
Quem não precisa ser internado?
A mortalidade de HDB é menor do que 1% e a maioria dos pacientes é manejado sem a necessidade de internação.
O ACG sugere a utilização de ferramentas de estratificação de risco com o objetivo de identificar pacientes de baixo risco que não precisariam ser internados. Um escore de Oakland (tabela 1) menor ou igual a 8 se correlaciona bem com uma alta segura , com 95% de probabilidade de não acontecerem intercorrências relacionadas ao sangramento em 28 dias.

Escores não devem substituir a impressão clínica. Apesar de alguns pacientes terem altas seguras, a oportunidade de alguns diagnósticos pode ser atrasada, como nos casos de câncer colorretal e doença inflamatória intestinal.
Como fazer o manejo específico da HDB?
Dois exames principais podem ser utilizados para diagnóstico: colonoscopia e angiotomografia (angioTC).
A principal função da colonoscopia é identificar a causa e excluir neoplasias. Apesar do poder diagnóstico, a capacidade da colonoscopia de conter sangramentos é baixa. A hemostasia dos exames realizados em até 24 horas chega a no máximo 20%. Por isso, o ACG faz uma recomendação forte com moderada qualidade de evidência que pacientes com HDB não devem realizar colonoscopia de urgência, em até 24 horas, por não haver redução de ressangramento ou mortalidade. Essa recomendação mudou em relação às últimas diretrizes. Uma exceção a ser considerada é o sangramento pós polipectomia.
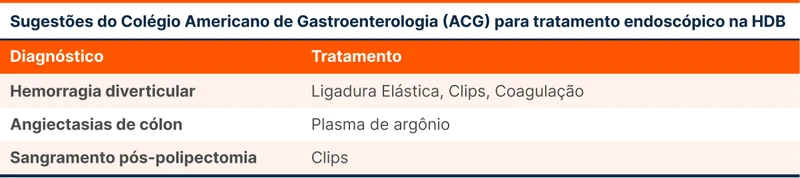
A tabela 2 traz as sugestões do tratamento endoscópico recomendado, cabendo ao endoscopista a definição da melhor técnica.
O preparo do cólon recomendado é com soluções de polietilenoglicol (PEG). A recomendação clássica é o uso de 4 a 6 litros de PEG em 3 a 4 horas até que as evacuações fiquem livres de sangue e fezes, mas outros protocolos podem ser utilizados. Os especialistas citam que os riscos desse preparo são semelhantes aos de pacientes que não estão sangrando.
A angioTC é um exame não invasivo que consegue identificar sangramentos e delimitar a anatomia vascular. O ideal é que a angioTC seja realizada dentro de 4 horas do início da hematoquezia. Quando a angiotomografia encontra o foco do sangramento, o controle é feito por embolização arterial via radiointervenção imediatamente após o exame ou por colonoscopia.
Faltam melhores estudos comparando as estratégias diagnósticas e não há evidência atual de diferença de mortalidade entre elas. A escolha é baseada em estudos retrospectivos e experiência do local. A diretriz sugere que a colonoscopia seja realizada nos pacientes estáveis e a angiotomografia nos pacientes instáveis ou com sangramento vigente (vide fluxograma 1).
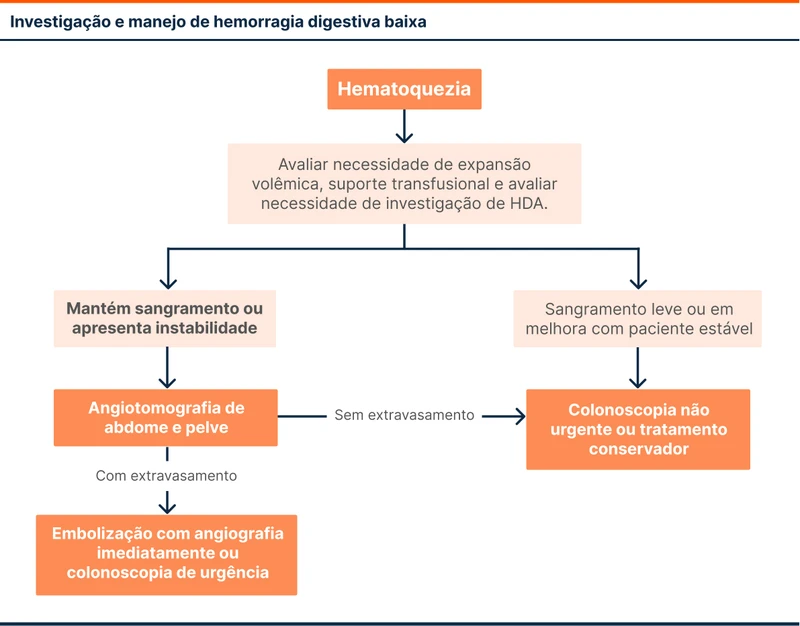
O manejo conservador, sem a realização da colonoscopia ou embolização, pode ser considerado em pacientes que pararam de sangrar e que têm confirmados a presença de doença diverticular e ausência de câncer, em exame realizado no último ano. Uma angioTC negativa tem valor para indicar estratégias conservadoras pois 80% desses pacientes melhoram sem necessidade de endoscopia ou rádio intervenção.
A cintilografia com hemácias marcadas vem sendo cada vez menos solicitada. O exame é demorado e não consegue localizar precisamente a região do sangramento.
A cirurgia é necessária em apenas 0,2% dos casos, quando há falha na hemostasia com angiografia ou colonoscopia. A maioria das cirurgias são de hemicolectomia e apenas 15% de todas as cirurgias são colectomia total.


