Hiperaldosteronismo Primário
Em abril de 2022, o British Medical Journal (BMJ) lançou uma revisão sobre hiperaldosteronismo primário (HP). Indo da investigação ao tratamento, o artigo coloca em pauta o porquê de ser uma condição tão subdiagnosticada [1]. Trazemos aqui os principais pontos.
O que é hiperaldosteronismo primário?
O hiperaldosteronismo primário (HP) é caracterizado pela produção inapropriadamente alta de aldosterona, independente dos níveis de renina. Isso aumenta a reabsorção de sódio e causa hipertensão.
Estima-se uma prevalência de HP de 3,2 a 12,7% entre pacientes com diagnóstico de hipertensão na atenção primária [2], sendo maior em pacientes com hipertensão resistente - chegando até 22% [3]. Apesar da alta prevalência, HP é uma doença subdiagnosticada.

Registre-se e ganhe 2 tópicos gratuitos da sua preferência!
Se você já é um membro do Guia TdC, faça o Login. Faça o login e conheça o Guia!Veja o que o Guia TdC te oferece:
- O Guia TdC é uma biblioteca de estudos bem selecionados resumidos em texto e em áudio, juntamente com todas as referências utilizadas. Tudo isso por um preço super acessível.
- Receba semanalmente as últimas recomendações e novidades com impacto direto na prática diária.
- Junte-se aos mais de 3.000 assinantes e atualize-se sem esforço.
Qual o motivo do subdiagnóstico?
Em geral, o rastreio de HP é indicado em pacientes com hipertensão resistente ou com hipertensão e hipocalemia. O subdiagnóstico pode ser decorrente dos seguintes motivos:
- Baixa taxa de rastreio em pacientes com hipertensão resistente - 2,1% em um registro americano [4].
- HP é mais comum em pacientes com hipertensão resistente, porém pode estar presente em qualquer faixa de hipertensão [5].
- Hipocalemia não é tão frequente quanto se pensa - apenas 9 a 37% dos casos apresentam hipocalemia.
- Apesar de sintomas como fraqueza muscular, cãibras, parestesias, palpitações e poliúria estarem associados, são infrequentes e inespecíficos. A maioria dos pacientes é assintomática.
Pacientes com HP, quando comparados com pacientes com hipertensão essencial, apresentam maior risco de AVC, doença coronariana, fibrilação atrial e insuficiência cardíaca, mesmo quando ajustado para os níveis de hipertensão [6].
Como fazer o diagnóstico?
O rastreio deve ser considerado em:
- Hipertensão resistente - Controle pressórico inadequado com 3 medicamentos, sendo um deles um diurético.
- Hipertensão moderada a grave - ≥140/90 mmHg.
- Hipertensão com massa adrenal incidental.
- Hipertensão com hipocalemia.
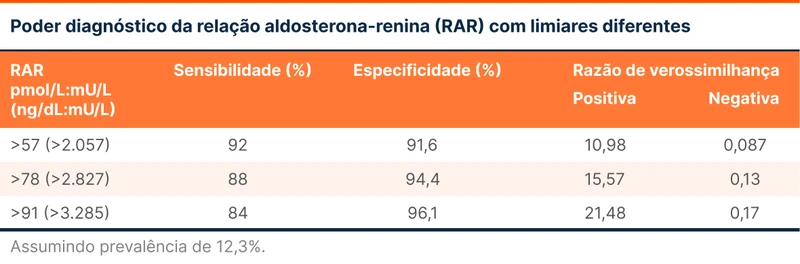
O teste de rastreio de escolha é a relação aldosterona-renina plasmática (RAR), sendo que a sensibilidade e especificidade variam conforme alguns limiares (tabela 1).
Alguns cuidados que devemos tomar para a realização da RAR:
- RAR é afetada pela maioria dos anti-hipertensivos utilizados rotineiramente (tabela 2).
- A coleta deve ser realizada 2 horas após despertar e ter deambulado, com ao menos 15 minutos de descanso antes da coleta.
- Doença renal crônica tende a reduzir os níveis de renina, podendo falsear a RAR para cima.
- Doença renovascular tende a aumentar a renina plasmática, tendendo a falsear a RAR para baixo.
- Manter potássio entre 4,0-5,0 mmol/L, visto que hipocalemia por ocasionar RAR falso-negativo.

Apesar de um teste imperfeito, duas medidas negativas em dias diferentes ajudam a excluir HP.
Em casos de RAR positivo, deve-se preferir encaminhar ao especialista para testes confirmatórios (teste de infusão com salina, teste de supressão com fludrocortisona ou teste de sobrecarga oral de sódio).
Como manejar?
Após o diagnóstico, precisamos estabelecer a etiologia. As duas principais causas de HP são: adenoma de adrenal e hiperplasia adrenal.
Os testes de escolha para investigação são tomografia computadorizada de adrenal (avalia a presença de adenoma) e amostra venosa adrenal (avalia se a produção de aldosterona é uni ou bilateral).
Em casos de adenomas produtores de aldosterona unilaterais, a terapia de escolha é a adrenalectomia unilateral.
Em pacientes com doença adrenal bilateral, a terapia de escolha são os antagonistas dos receptores de mineralocorticoides. A medicação de escolha é a espironolactona, iniciada em doses de 12,5 a 25mg, com doses máximas de 100 a 400mg por dia [7]. Em casos de efeitos colaterais importantes (ginecomastia, mastodinia, redução da libído, alterações menstruais), pode-se optar pela eplerenona.
A meta de tratamento é a normalização da pressão, da hipocalemia e da concentração de renina.